Tautomer Search
Many medicinally relevant molecules exist as multiple tautomers, and understanding which tautomer predominates can be key to subsequent computational tasks: for instance, Hu et al found in 2016 that correct assignment of tautomeric state dramatically improved relative binding affinity predictions. Rowan's tautomer prediction workflow uses machine-learned interatomic potentials to enable fast and minimally empirical prediction of the relative stability of different tautomers.
How it works
Rowan's tautomer prediction workflow begins by using the tautomer enumeration functions in RDKit to generate all possible tautomers. The initial tautomer geometry is quickly optimized with GFN2-xTB, and a single-point energy is computed using the AIMNet2 model trained on ωB97M-D3BJ/def2-TZVPP training data, henceforth abbreviated as AIMNet2. Since AIMNet2 does not take solvation into account, ∆G_solv is computed from a single-point GFN2-xTB calculation with the CPCM-X implicit water model.
Rowan next discards all tautomers that are predicted to be extremely high in energy (above a mode-specific cutoff). For the remaining "significant" tautomers, conformers are generated using the ETKDG v2 algorithm and optimized using the MMFF94 forcefield. After removing redundant geometries, low-energy conformers are then optimized using GFN2-xTB, single-point energies are calculated using AIMNet2, and the lowest-energy conformers undergo full optimization with AIMNet2 to generate a final energy for each conformer, including a CPCM-X solvent correction. The final energy values for each tautomer are computed as the Boltzmann-weighed average of the individual conformer energies.
Accuracy
On the aqueous subset of the TautoBase benchmark set, Rowan's tautomer workflow displays a mean absolute error of 2.10 kcal/mol and a root mean squared error of 2.99 kcal/mol. This is comparable to the performance of high-level quantum chemical methods reported by Chodera and co-workers: B3LYP/cc-pVTZ/SMD(water) was reported to give an RMSE of 3.1 kcal/mol vs. TautoBase (on a slightly smaller subset).
A more relevant benchmark for real-world usage is classification accuracy—how much of the time can Rowan predict the correct lowest-energy tautomer? On the full dataset, Rowan predicts the correct tautomer 89% of the time. Some of these comparisons are not particularly challenging: for compounds with an experimental ∆∆G of less than 3 kcal/mol (shown in red), Rowan is still correct 77% of the time.
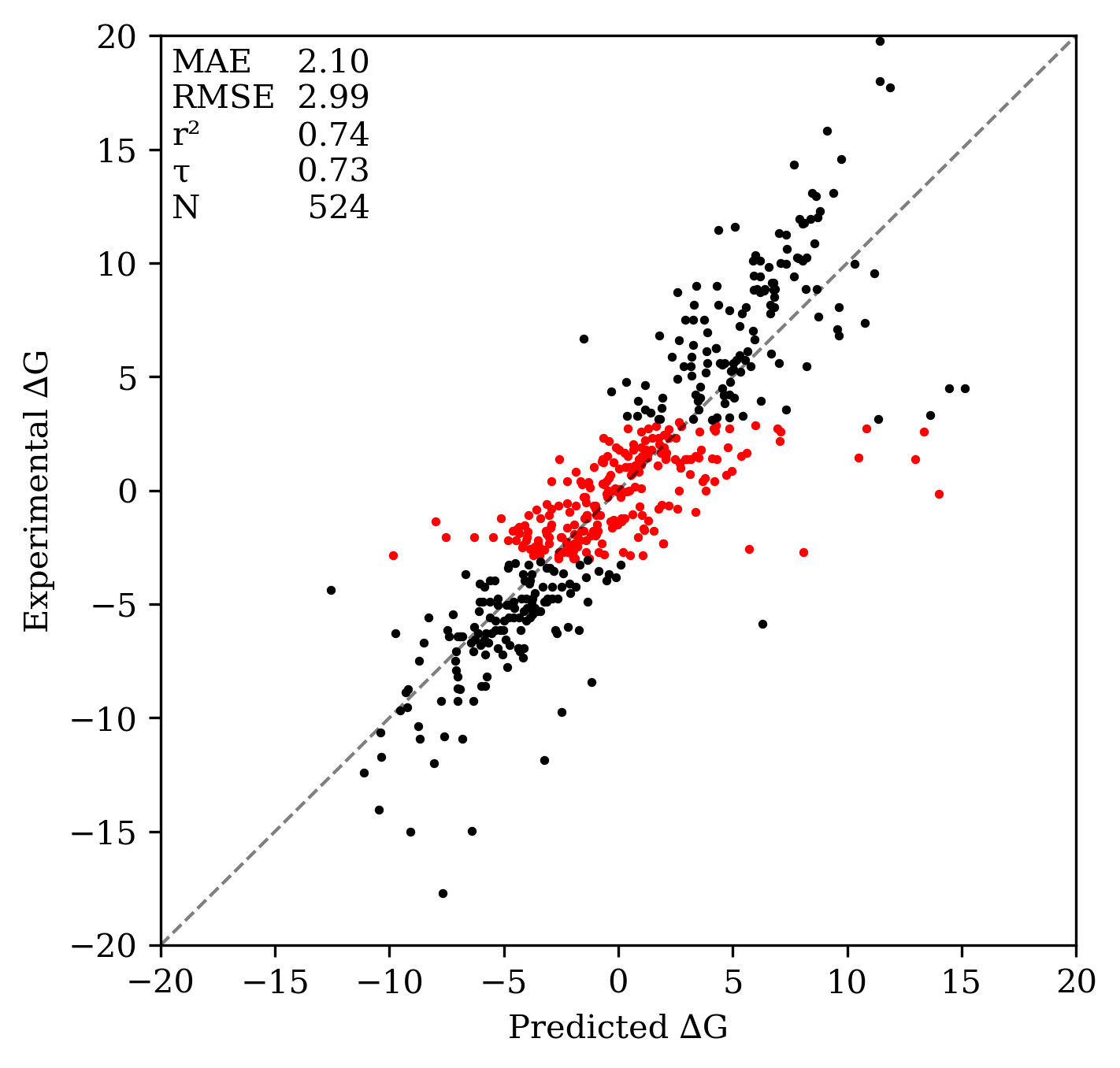
Rowan's tautomer search workflow's performance on TautoBase
Modes
Rowan's tautomer workflow can be run in three modes: careful, rapid, or reckless. Here's what selecting each mode tunes:
| Mode | Careful | Rapid | Reckless |
|---|---|---|---|
| number of initial conformations | 250 | 100 | 50 |
| initial energy cutoff (kcal/mol) | 15 | 10 | 5 |
| RMSD similarity cutoff (Å) | 0.10 | 0.25 | 0.50 |
| max number of conformers (xTB) | 20 | 10 | 3 |
| final energy cutoff (kcal/mol) | 5 | 5 | 3 |
| max number of conformers (AIMNet2) | 10 | 3 | 1 |