Hydrogen-Bond-Basicity Prediction
Understanding and controlling the strength of hydrogen bonds is key to modulating a molecule's interactions and physicochemical properties. Rowan's hydrogen-bond-basicity prediction workflow uses neural network potentials, low-cost quantum chemical methods, and molecular electrostatic potential calculations to efficiently and robustly predict the strength of different hydrogen-bond acceptors.
How it works
Rowan's hydrogen-bond-basicity workflow begins by using the ETKDG algorithm to run a conformer search on the input molecule. After initial MMFF94 optimization, we filter the resulting conformational ensemble using the CREST screening protocol and GFN2-xTB energies to de-duplicate structures and remove high-energy conformers. We use a 2% rotational constant threshold, a 0.25 Å RMSD similarity threshold, and a 50 kcal/mol energy cutoff window for CREST screening. We score & optimize the output conformers with the AIMNet2 neural network potential.
We then locate electrostatic potential minima in the region of each lone pair of the lowest energy conformer, using the low-cost r2SCAN-3c composite density-functional-theory method. pKBHX values are predicted by linearly scaling Vmin, the value of the molecular electrostatic potential at the minimum.
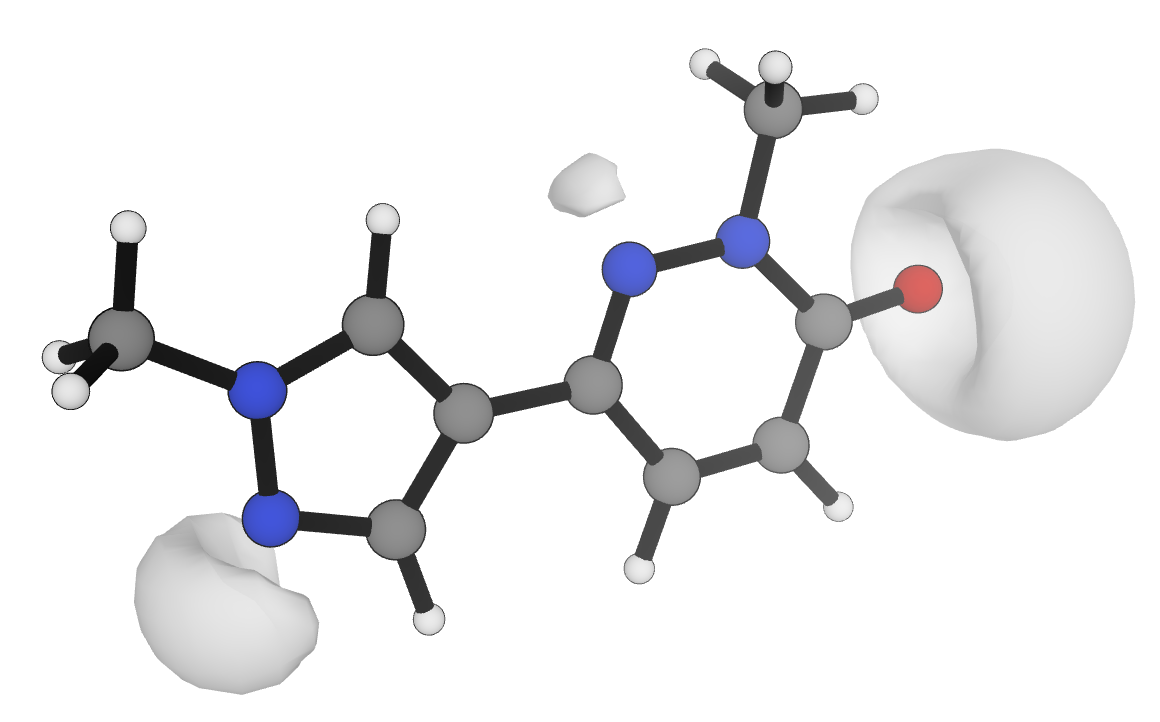
Visualization of the −0.04 electrostatic potential isosurface at the r2SCAN-3c level of theory for an example drug-like molecule.
To find values for the scaling constants, we performed Levenberg–Marquardt least-squares fitting to a database of experimentally measured pKBHX values, predicting the measured per-molecule pKBHX values by combining the predicted pKBHX values from each distinct electrostatic potential minimum (up to three per hydrogen-bond acceptor). Here are the scaling constants found through this procedure:
| Functional Group | Number | Slope () | Intercept | MAE | RMSE |
|---|---|---|---|---|---|
| Amine | 171 | -34.4386 | -1.4884 | 0.212 | 0.324 |
| Aromatic N | 71 | -52.8126 | -3.1376 | 0.113 | 0.150 |
| Imine | 28 | -48.4007 | -2.3309 | 0.180 | 0.236 |
| Nitrile | 28 | -50.1167 | -3.2273 | 0.144 | 0.198 |
| N-oxide | 16 | -74.3261 | -4.4159 | 0.455 | 0.589 |
| Chalcogen oxide | 17 | -47.7009 | -2.2794 | 0.186 | 0.224 |
| Pnictogen oxide | 16 | -61.1141 | -3.3839 | 0.437 | 0.549 |
| Carbonyl | 128 | -57.2911 | -3.5271 | 0.160 | 0.208 |
| Ether/hydroxyl | 99 | -35.9245 | -2.0338 | 0.188 | 0.239 |
| Thiocarbonyl | 10 | -51.8837 | -2.2649 | 0.330 | 0.384 |
| Divalent S | 17 | -39.1666 | -2.1243 | 0.086 | 0.127 |
| Aromatic O | 11 | -35.9245 | -2.0338 | 0.125 | 0.158 |
| Fluorine | 23 | -16.4441 | -1.2540 | 0.202 | 0.276 |
| Total | 434 | 0.188 | 0.270 |
For a more detailed discussion of outliers and potential sources of error, see the preprint.