Free-Energy Perturbation
This is a guide about how to run RBFE calculations through Rowan.
What you need to run FEP
To run Rowan FEP, you'll need a bound protein–ligand pose. Ideally, this would come from structural biology data (e.g. X-ray crystallography), but if necessary docking or co-folding can be used; as FEP will inherit structural errors from the input structure, use of these methods may result in lower accuracy results. You'll also need a list of analogues in SMILES format. (If you already have bound 3D poses for all the analogues, feel free to skip Step 1 and proceed straight to Step 2.)
A few notes on choosing analogues: small perturbations lead to faster calculations, but larger perturbations like scaffold hops can also be handled. At a minimum, there should be substantial structural overlap between all compounds to enable the alchemical perturbation. (If you're interested in comparing totally unrelated compounds, RBFE is probably the wrong technique; consider using endpoint-based methods or ABFE. We don't yet have Rowan-native solutions here, but hope to add these in the future.)
At this point, Rowan does not have capabilities to automatically suggest nearby analogues; if you don't know which analogues to pick, consider open-source chemical replacement models like CReM, machine-learned generative models, or old-fashioned chemical intuition.
(For the remainder of this tutorial, we'll be looking at modifications to this Rock2 system from the Uni-FEP benchmark set.)
Step 1: Generating aligned poses
To generate aligned poses for the analogues, select Rowan's "Analogue docking" workflow and input the protein, the bound reference pose, and the analogues in question.
You may need to clean up the protein structure, particularly if it's experimental. Rowan will display an error if the protein requires additional preparation; here, the protein is missing hydrogens:
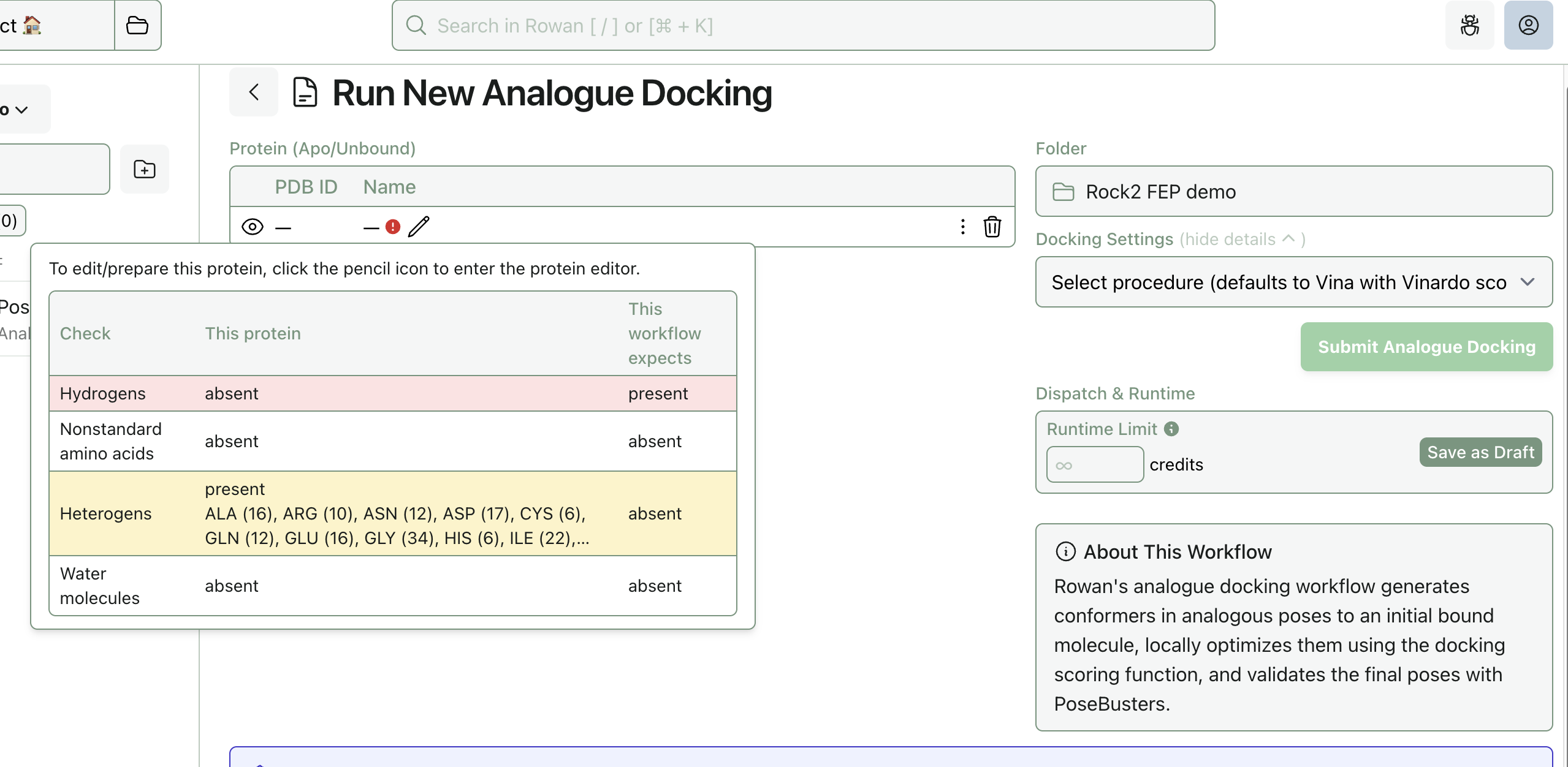
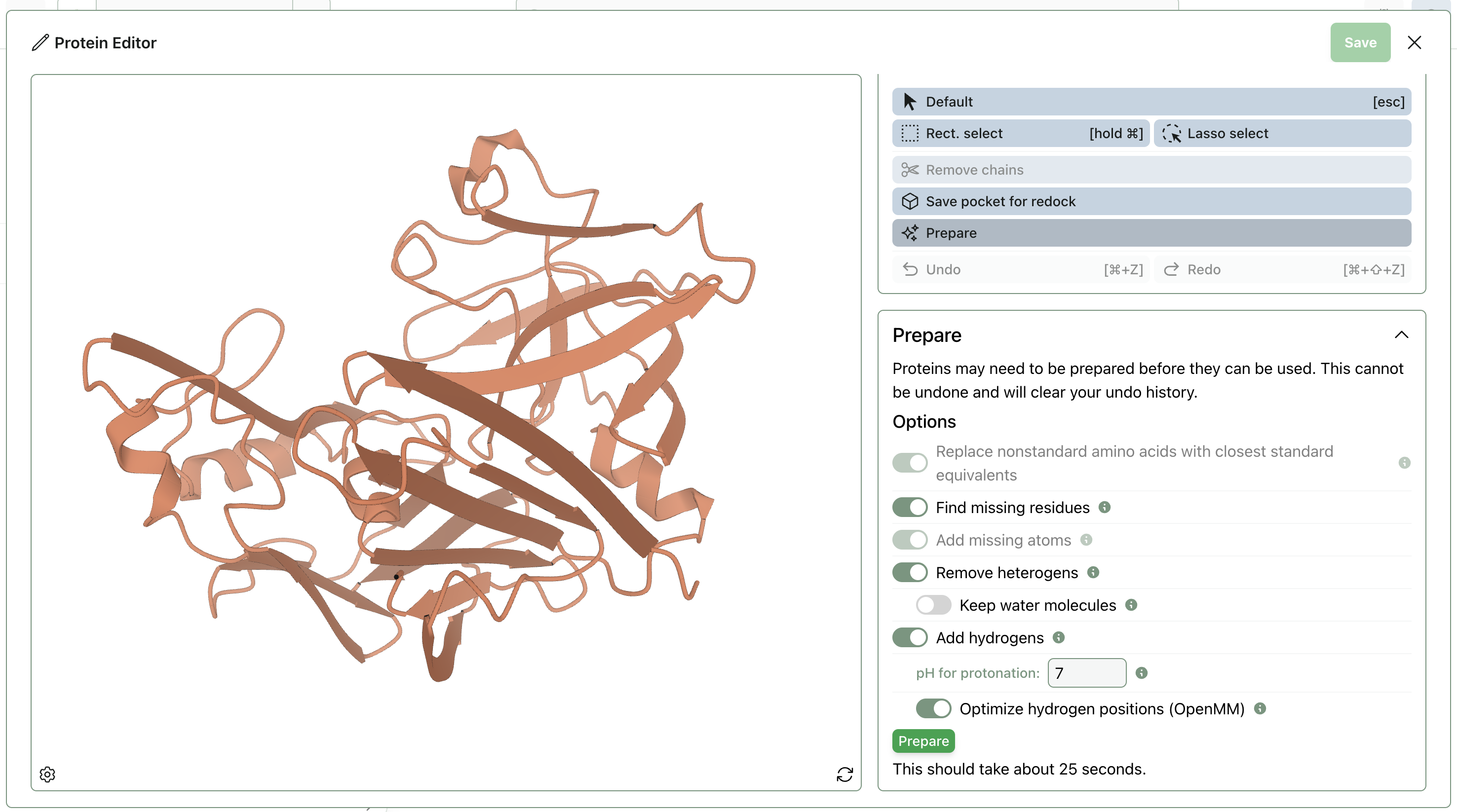
To prepare the protein, click the "Edit" button (pencil icon) to enter the "Protein editor" view. There, you can remove chains and run protein-preparation routines.
Once the protein is properly prepared, you can upload the bound reference pose and input the analogues as SMILES. The structure of each molecule can be visualized by clicking the eye icon on each row.
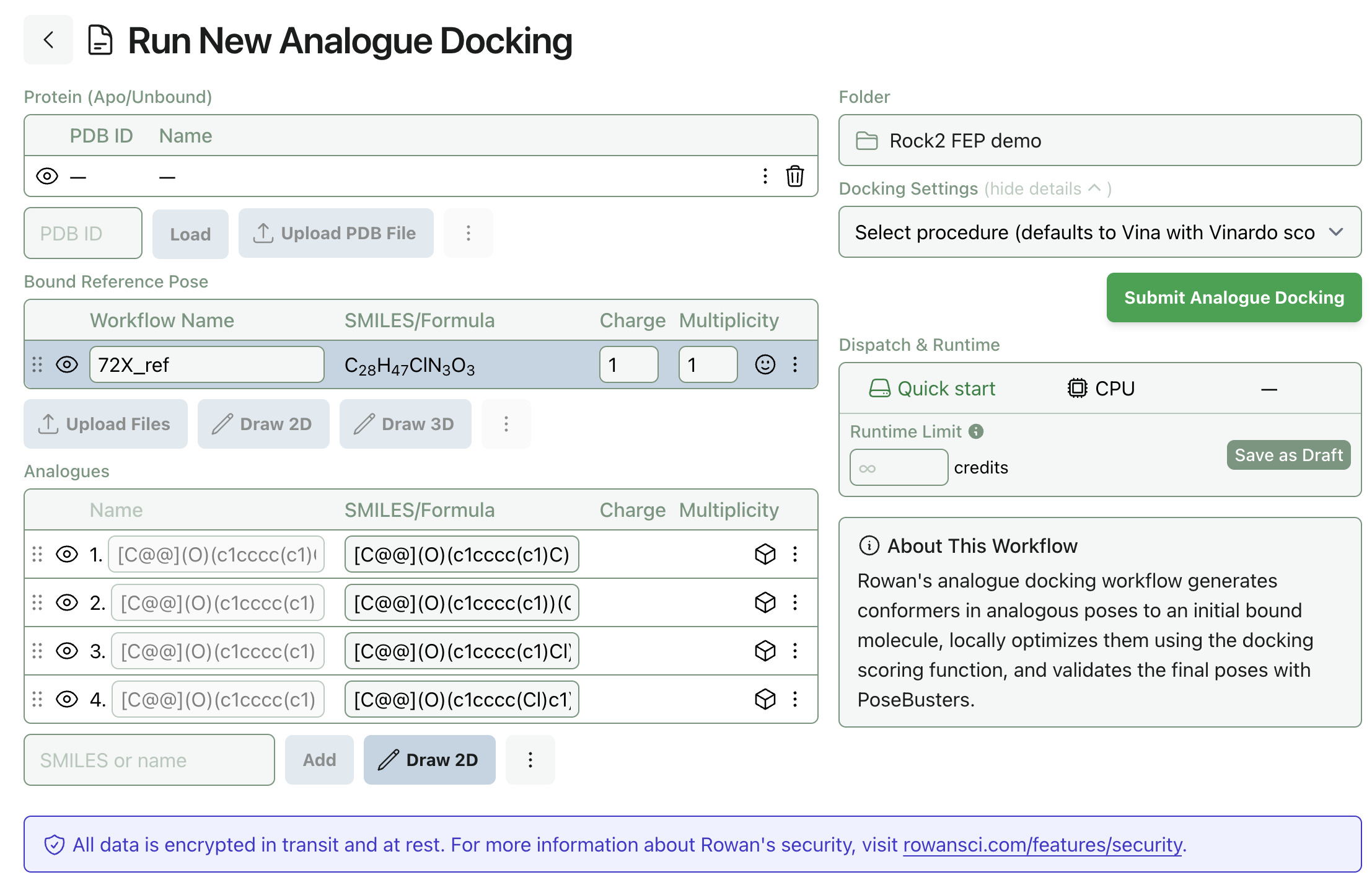
Once the input has been correctly prepared, click "Submit analogue docking" to start the workflow. (Note that analogue docking does not, by default, include the reference pose in the list of analogues—you may wish to manually add the pose's SMILES representation here.)
The workflow will take a few minutes to run per analogue. Once complete, you can view different poses for each ligand, with their corresponding docking scores and MCS RMSD values, and overlay different analogues to see how their binding poses compare.
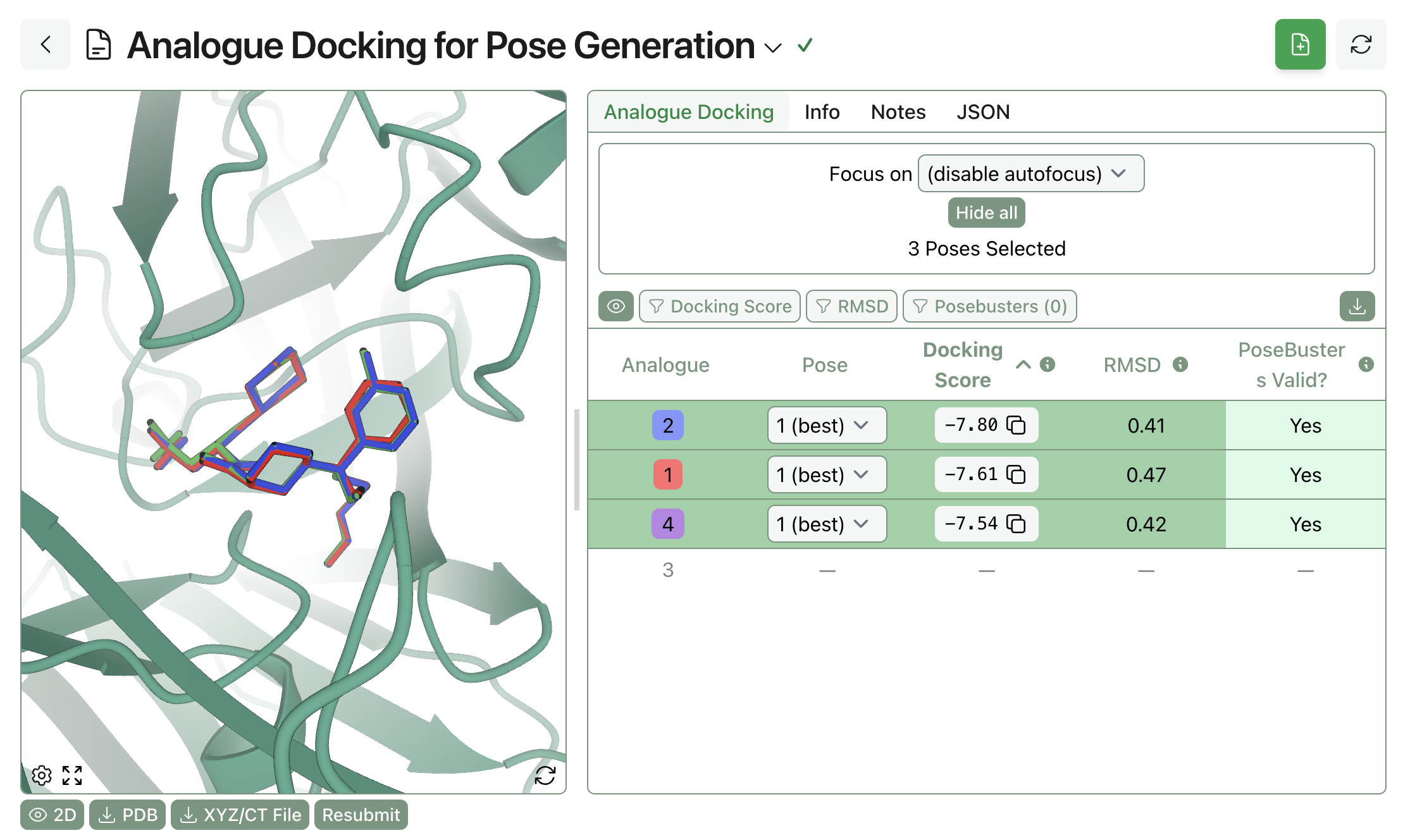
Particularly large perturbations, like compound #3 above, may fail via analogue docking. In these cases, separate docking calculations can be run to generate initial poses.
Step 2: Build RBFE graph
With poses in hand, the next step is to build the graph that shows which alchemical perturbations will actually be computed. To start, select all poses (that you want to submit) and click "Resubmit" below the protein viewer. Then, navigate down to "Resubmit as RBFE graph workflow." On the "RBFE graph workflow" submission page, you can add additional poses (e.g. from other docking runs), manually edit poses, and add per-compound identifiers.
The right side of the page allows you to control how the RBFE graph is built. Here's what the options mean:
- Graph mode determines how the graph is built. At present, there are two options. The default "greedy" method will generate a standard redundant RBFE graph that connects similar compounds to each other. In contrast, the "star map" method will put one compound in the center and connect all the other compounds via single edges (which is faster but typically less accurate).
- The Scoring method controls how ligand similarity is determined for greedy graph construction. "Jaccard" uses Jaccard/Tanimoto similarity between ligand structures, while "Dummy atom" counts the number of dummy atoms that will be required. The default "Best" method builds graphs with both methods and chooses the one with the lower mean dummy-atom count.
- K min cut determines the redundancy of the graph (for greedy graph construction): the default value of 3 means that every ligand will have at least 3 edges connecting it to other ligands. Larger values will lead to more redundancy in the graph, while smaller values will lead to faster calculations.
If you're new to FEP, we recommend choosing the "greedy" graph-construction mode with default settings: this will generally work well and give robust results. If you want things to run slightly faster, you can reduce k min cut to 2. If you're in a hurry and willing to compromise on result quality, use the "star map" mode and choose a good central compound.
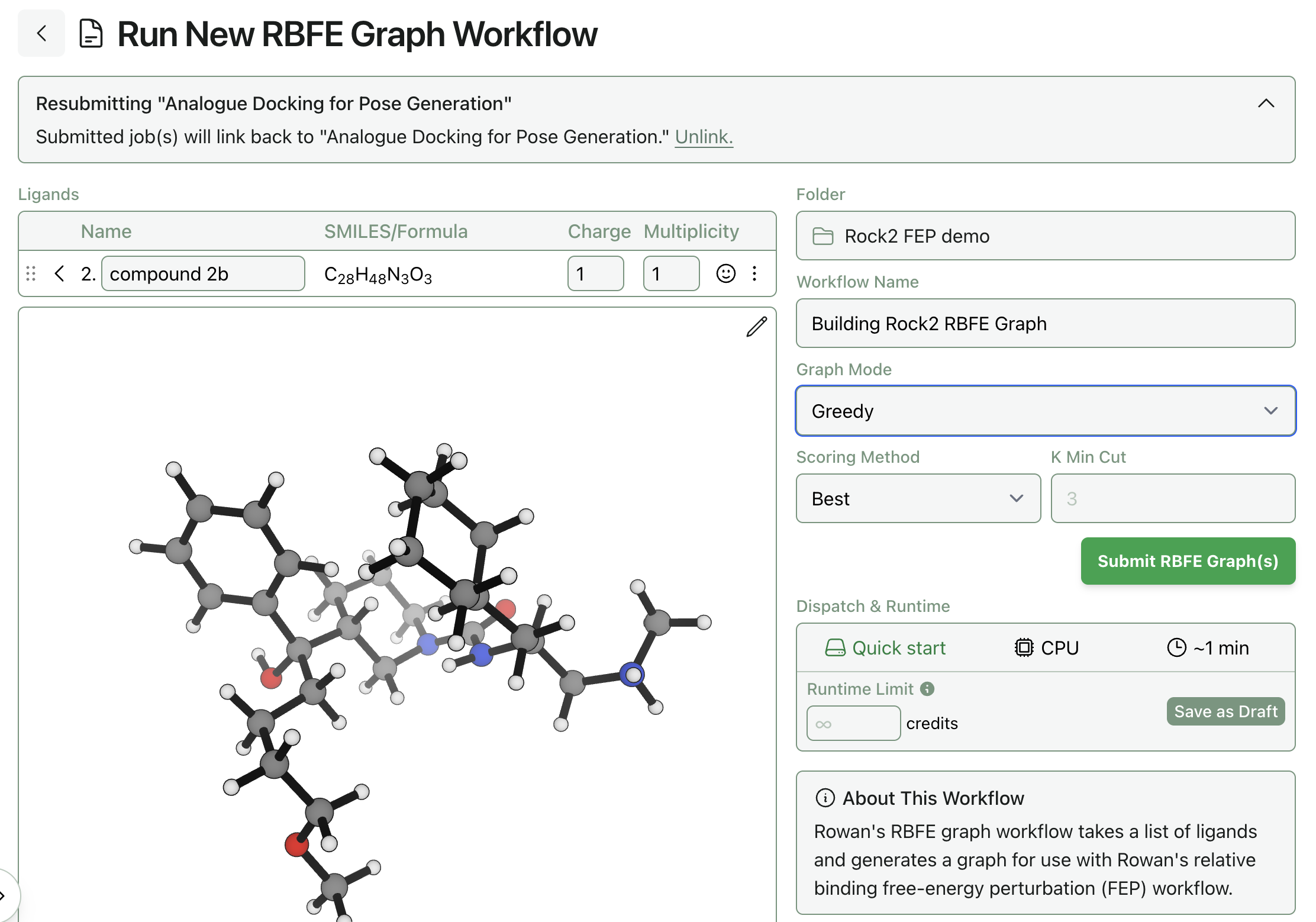
Once the poses and settings are all correct, click "Submit RBFE graph." This workflow will typically run quite quickly. Here's what the output of a simple star-map FEP graph looks like:
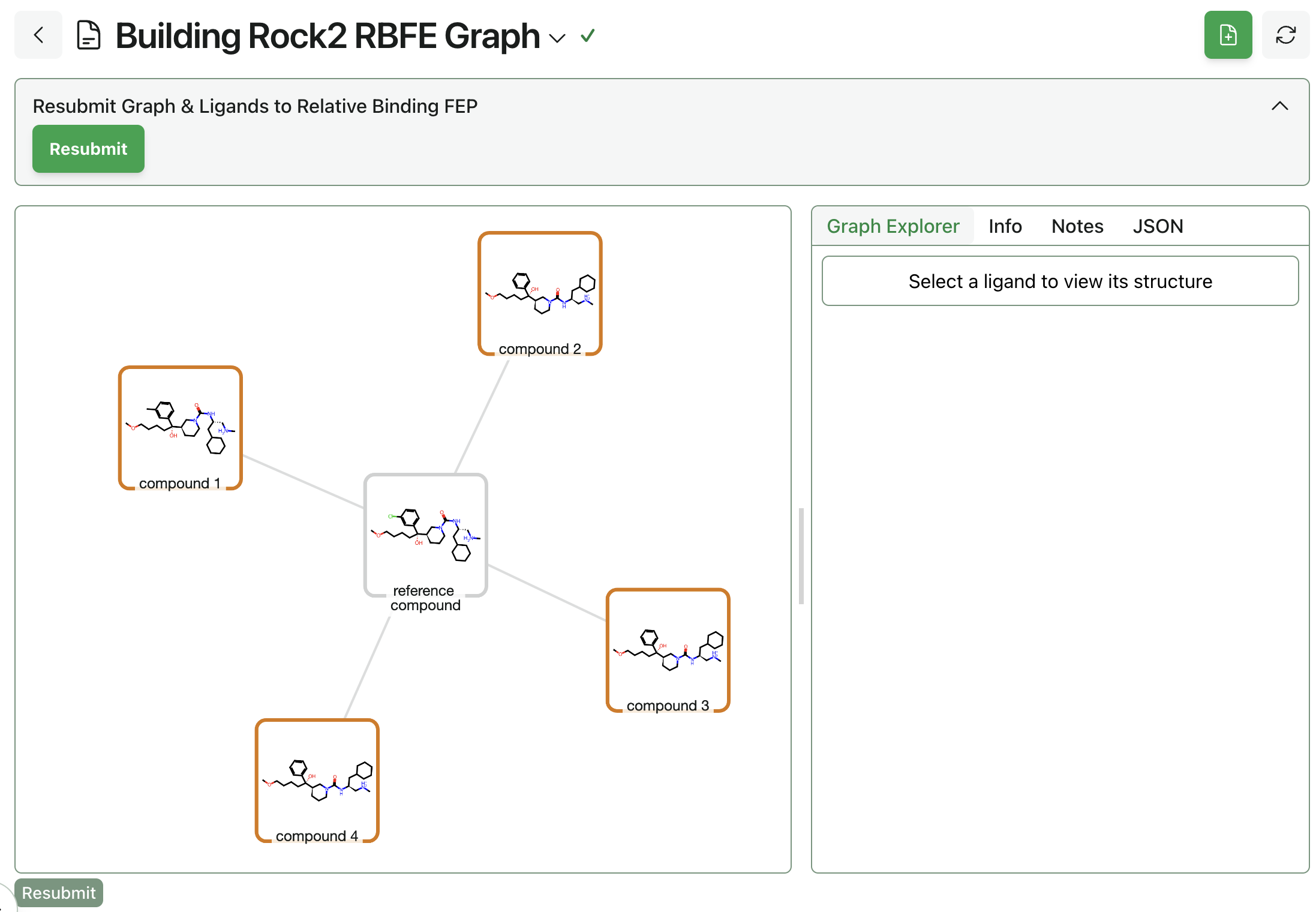
Step 3: Run FEP
Once the graph has been built, click "Resubmit" at the top to go to the FEP submission screen. If you already ran analogue docking, the protein should already be associated with the workflow; if not, you'll need to upload it now (and potentially run the protein preparation steps above).
The FEP submission screen will let you see the protein, the perturbation graph, and the settings. If you want to manually add or remove edges, you can do so by clicking the pencil icon at the top right of the graph.
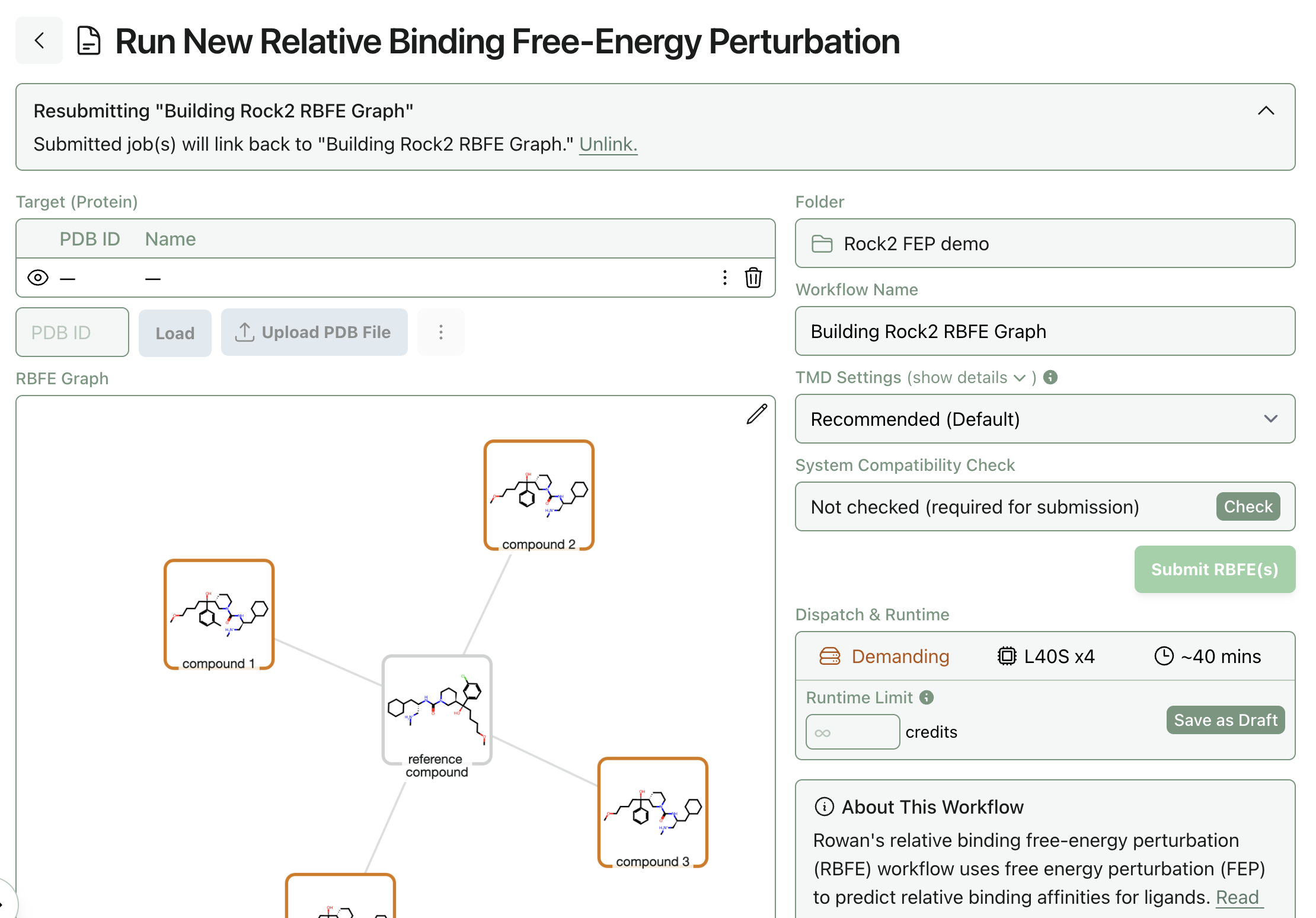
To adjust FEP settings, you can click "show details" in the TMD settings dropdown, which will expose all of the internal TMD settings.
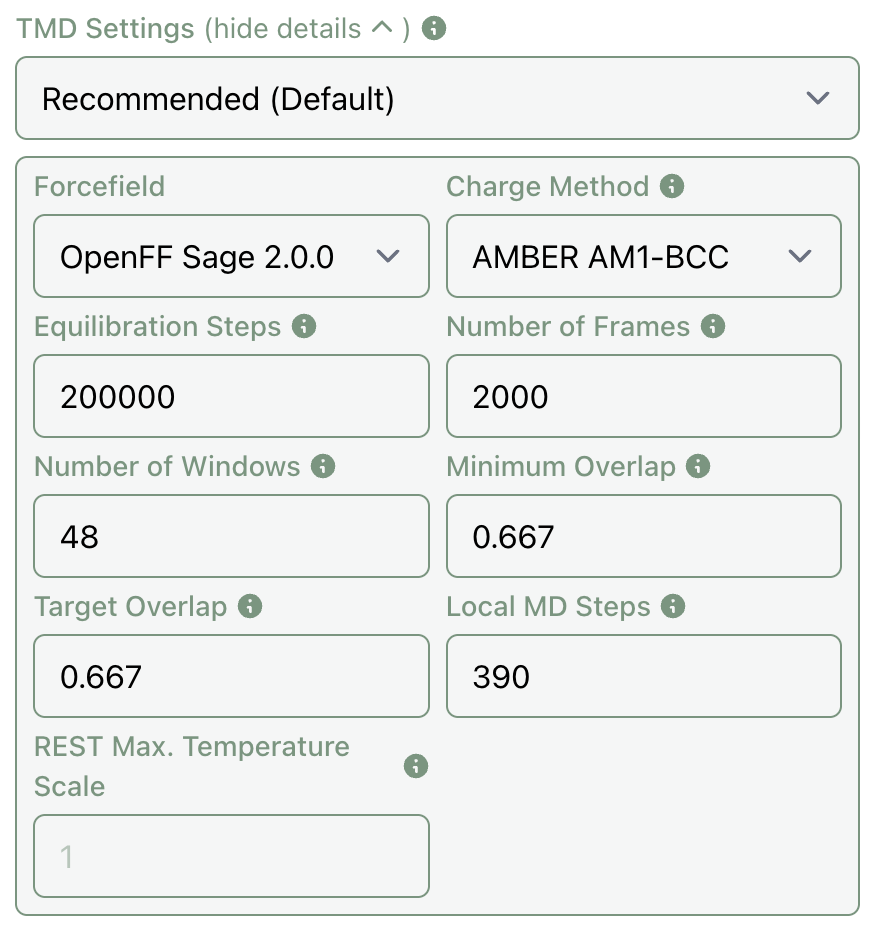
Here's what these settings mean:
- The forcefield determines which forcefield is used to parameterize the small-molecule ligand.
- The charge method controls how partial charges are assigned: either through semiempirical AM1-BCC calculations or via the NAGL graph neural network.
- Equilibration steps sets how long each trajectory will undergo equilibration before production runs commence. Since TMD uses a 2.5 fs timestep, the default of 200K steps corresponds to 0.5 ns of equilibration time.
- The number of frames controls how long the production trajectories will run for; each frame comprises 400 steps, so the default of 2000 frames corresponds to 2 ns of production time.
- The number of windows controls how many distinct alchemical windows are used for each perturbation. (TMD will reduce this number automatically for easy perturbations.)
- The minimum overlap dictates the minimum required overlap between adjacent alchemical windows, while the target overlap sets the desired overlap.
- The number of local MD steps controls TMD's local resampling approximation, which uses a cutoff radius approximation to lead to significantly faster simulations. This parameter sets how many local steps are taken per 400-step frame: the default value of 390 means that 390/400 will be local and 10/400 will be global. To disable local resampling, set this value to 0.
- Finally, the REST maximum temperature scale parameter sets the maximum temperature scaling factor for REST (replica exchange with solute tempering). Setting this value to 1 disables REST (the default), while increasing it will enable REST.
For first-time users, we recommend leaving most of these parameters as they are, as they've been found to work well for TMD through trial-and-error. The one exception is the charge-assignment method: AM1-BCC can get slow, so we recommend switching to NAGL for large molecules.
If slightly more control is desired, different preset settings can be accessed through the "TMD settings" dropdown. The "Fast" settings will reduce trajectory lengths and required overlap but might lead to increased error, while the "Rigorous" settings are identical to the default but without local resampling.
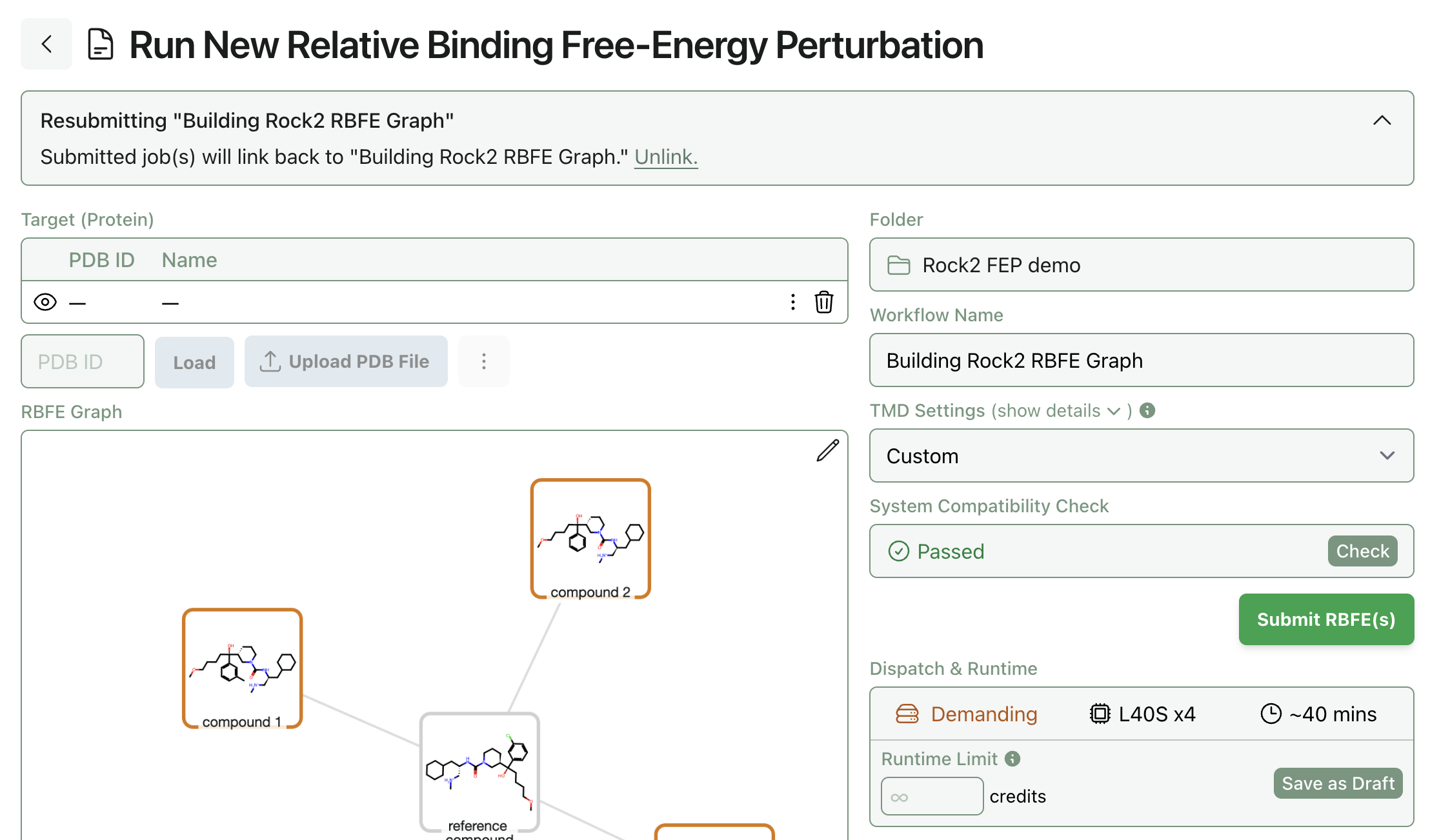
Before running FEP, you'll need to run the "System compatibility check" to verify that the protein can successfully be parameterized by TMD. This can occasionally be tricky: proteins prepared in other software sometimes clash with the TMD/OpenMM atom-naming scheme, and it can be necessary to remove all hydrogens (or just the invalid hydrogens) and add them back using the protein editor. Residue and atom names need to follow AMBER conventions, as Rowan FEP uses AMBER ff14SB for the protein.
Once the system compatibility check has passed, you can submit the RBFE workflow. RBFE workflows take a bit longer to start running than other Rowan workflows, since we have to allocate quite specialized hardware. You can select "Email when complete" to get an email notification when the job finishes.
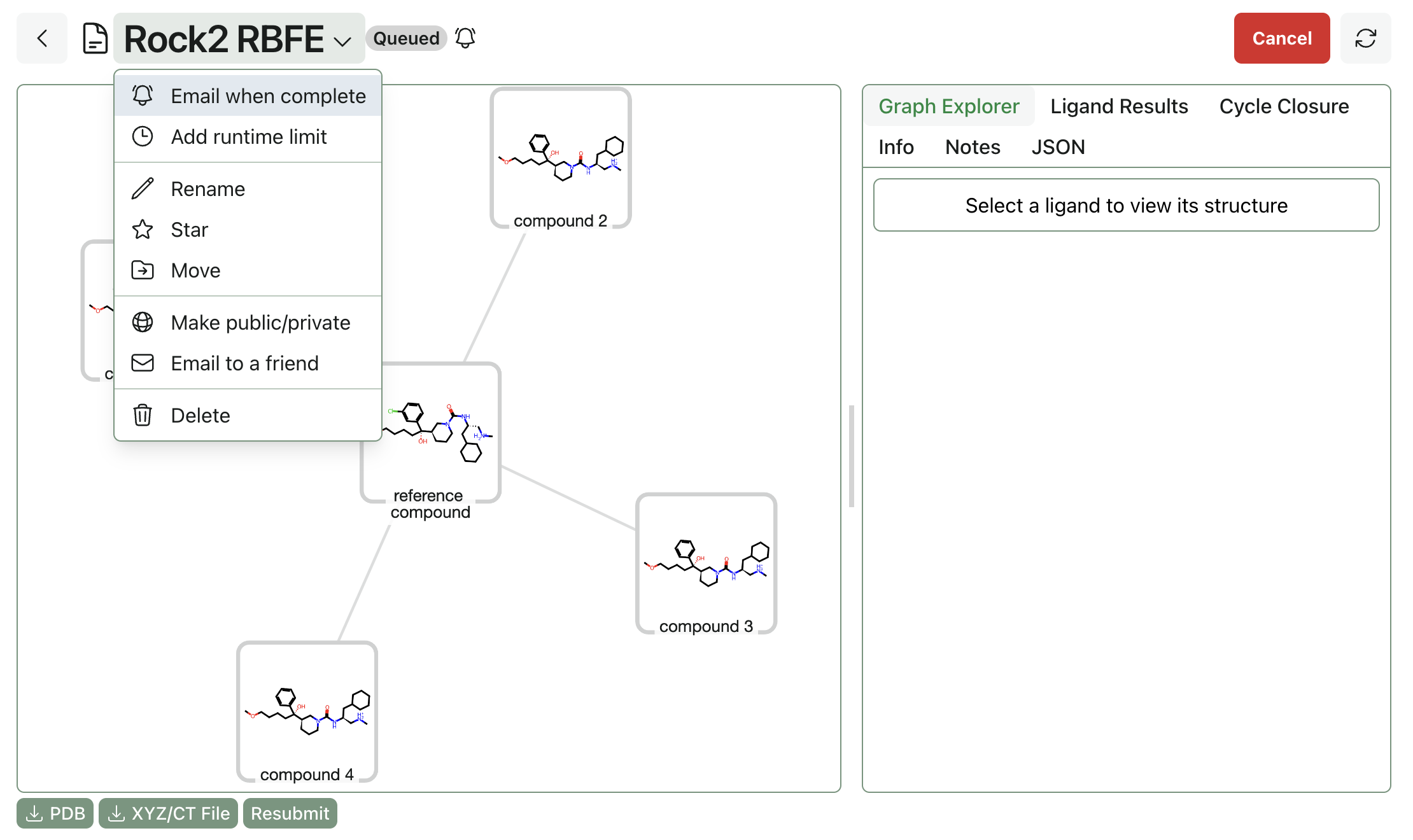
As the job runs, you'll be able to see legs start to complete. Per-edge ∆G values will be visible for completed legs.
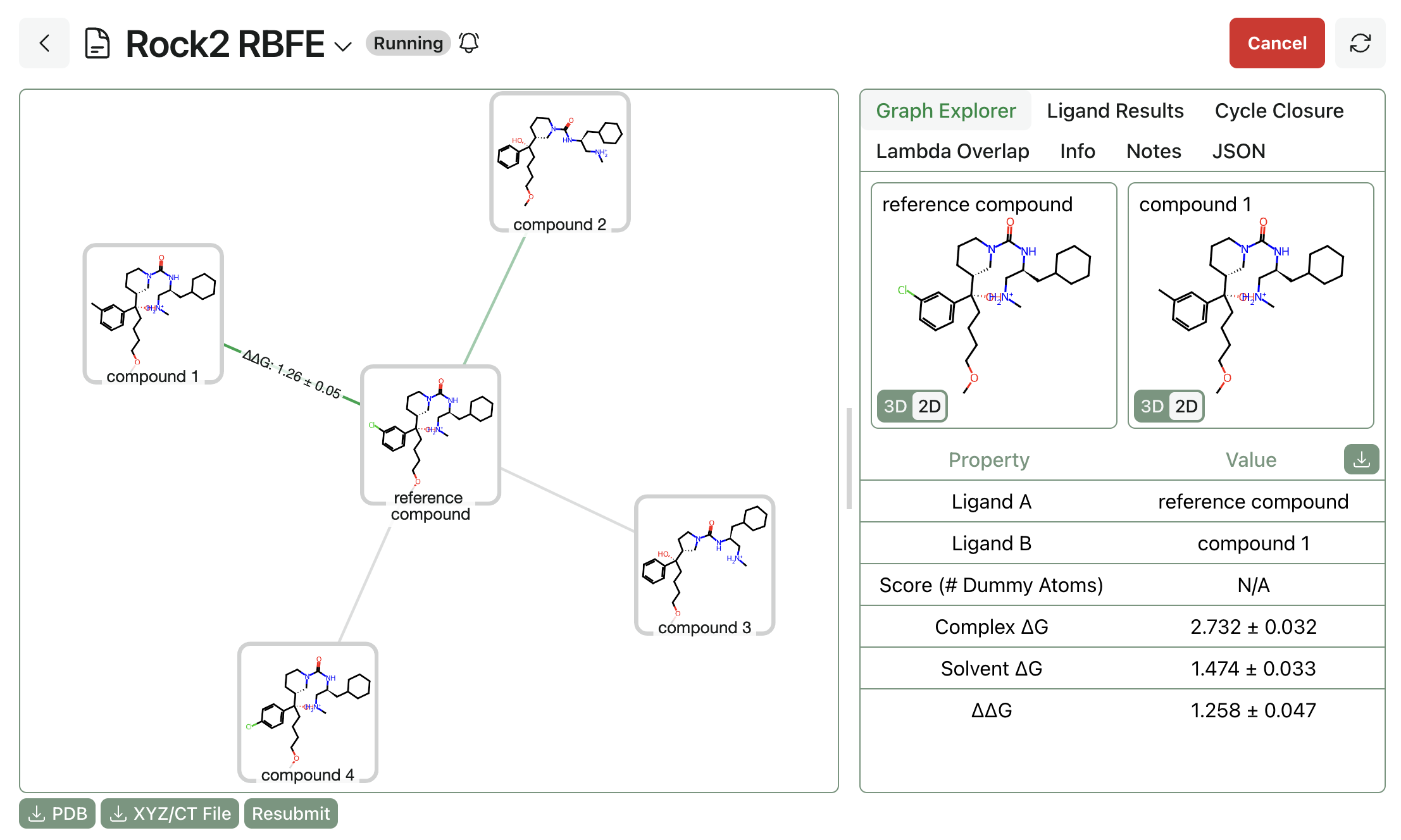
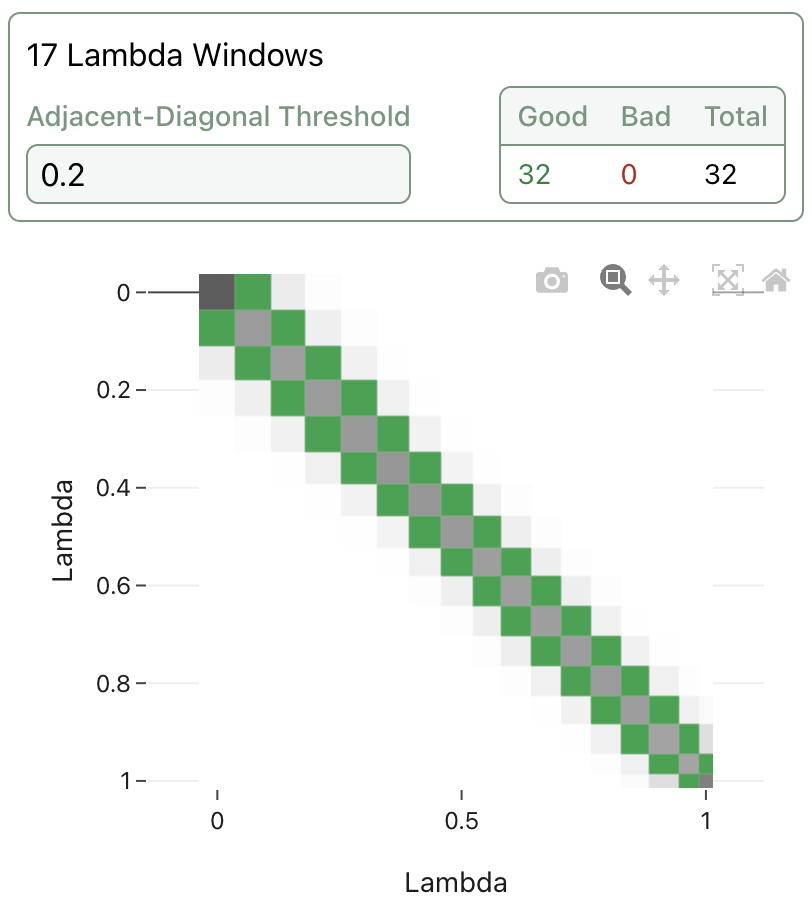
Lambda overlap values are also visible for completed legs. This gives insight into any potential sampling issues and allows users to see the outcome of TMD's adaptive lambda scheduling algorithm:
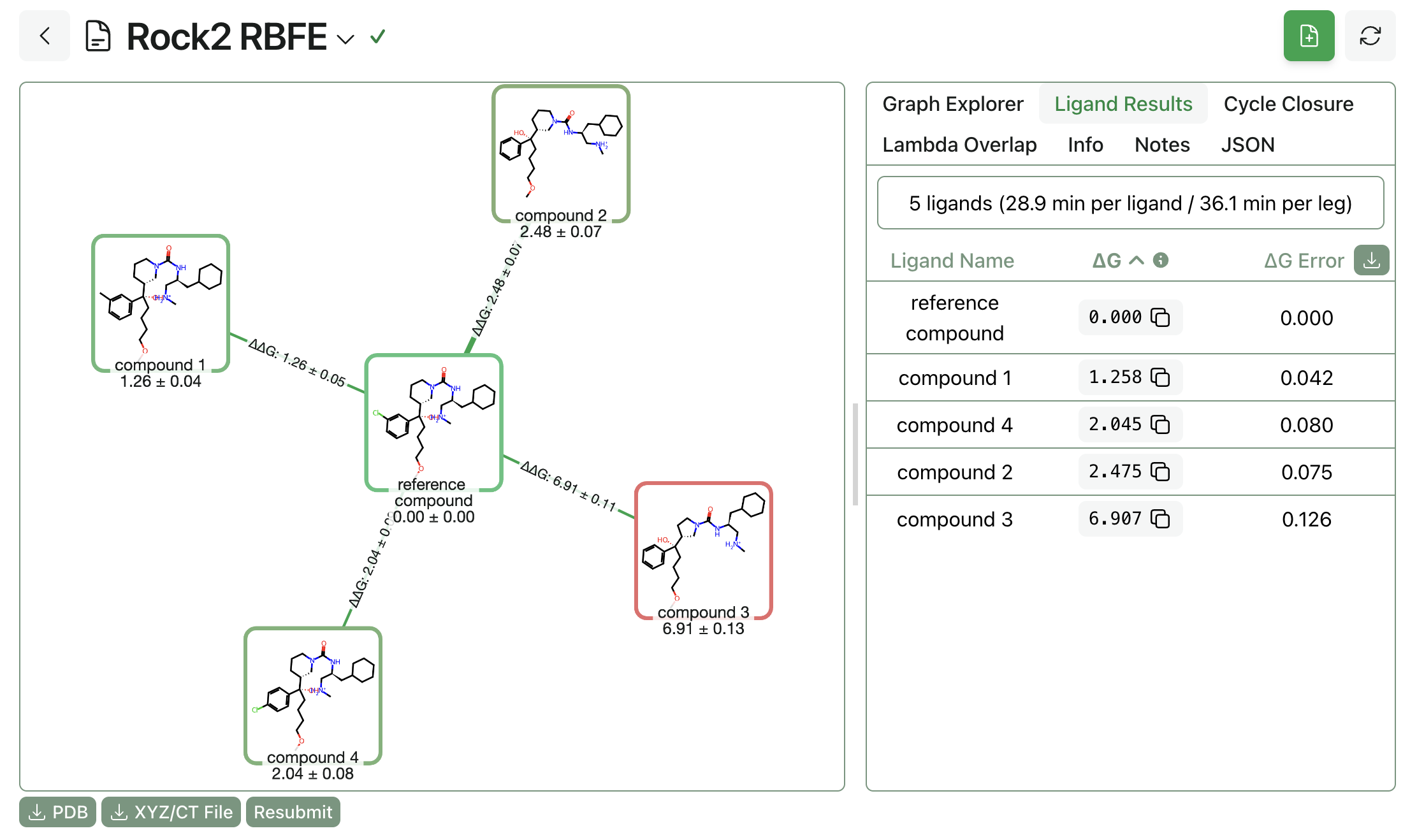
Overall ligand and timing results will be visible once the workflow completes. Here's the final list of ligand results from the example run; as expected, the reference compound outperforms the analogues.
Frequently asked questions
How do I compare these results to experimental data?
FEP returns relative binding affinity data, so absolute binding affinities aren't calculated. We recommend adding a constant such that the mean of the computed predictions matches the mean of the experimental predictions. After aligning the two datasets, standard statistical comparisons can be run: mean absolute error, root-mean-squared error, Pearson correlation, Spearman correlation, and so on.
Why is my calculation is taking longer than expected?
The length of FEP runs depends on many factors: the difficulty of the alchemical transformation, the size of the protein, the settings employed, and the quality of the input poses. Here are a few common reasons why a run might be slower than expected:
- AM1BCC charges are being used to parameterize large ligands. For ligands with more than 60 or 70 atoms, we recommend using NAGL charges since the semiempirical AM1BCC charge-assignment process can become very slow.
- The alchemical transformations are difficult. FEP legs that require large structural rearrangement, like adding bulky groups or dramatically changing conformations, will be difficult and slow to converge. In contrast, simple substitutions (e.g. Cl for Me) typically run quickly. FEP can be very useful for difficult transformations, but don't be surprised if they run slowly!
- The poses are poorly aligned. Poorly aligned poses make the transformations more difficult than they need to be. We recommend using Rowan's analogue docking workflow and excluding any poses predicted to bind via radically different poses, as they will make FEP much more challenging.
What engine powers Rowan FEP?
Rowan's FEP workflow is powered by TMD, a high-performance FEP engine maintained by Glysade. TMD is based in part on the Time Machine engine developed at Relay Therapeutics, but has been extensively rewritten and optimized to achieve maximum performance on modern GPU hardware. TMD also incorporates many algorithmic improvements to improve performance and accuracy, including local resampling, adaptive lambda scheduling and grand canonical Monte Carlo water sampling.
Can I combine ligands with different charges?
TMD does not support combining ligands with different charges; instead, split the ligands by charge state and compare them separately.
Can Rowan FEP handle membrane proteins?
Rowan does not support embedding proteins within membranes for FEP; anecdotally, it's sometimes fine to just ignore the membrane, but extra caution is certainly warranted.
Can Rowan FEP handle cofactors?
Rowan FEP does not yet support inclusion of small-molecule cofactors; we hope to add this functionality in the months to come. If you'd like to prioritize this development, reach out to our team.
Can Rowan FEP model covalent inhibitors?
Explicit bond-forming processes cannot be modeled through Rowan FEP, but the non-covalent portion of inhibitor binding can be modeled.